最好新建一個python環境,因為我發現conda安裝blast默認的是python==3.6.11,可能會不小心把你的python版本改掉…然後你寫好的代碼全die了……
conda create -n blast python==3.6.11
source activate blast
conda install -c bioconda blast
nr和uniprot是比較通用的數據庫:
ftp://ftp.ncbi.nlm.nih.gov/blast/db/
https://www.uniprot.org/downloads
1)nr是ncbi收集的目前所有微生物的蛋白序列,是用來計算氨基酸一般情況下的頻率的,160G
2)uniprot90根據相似性做了一個去冗余,所以比nr要小很多,56G
# 以uniprot90為例
wget ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref90/uniref90.fasta.gz # 下載
gzip -d uniref90.fasta.gz # 解壓
makeblastdb -in uniref90.fasta -parse_seqids -hash_index -dbtype prot # 編譯
解析完成後的樣子:

文件是這個樣子:(只截取了一部分)
我的初始文件是:
P00269.fasta是對單條蛋白質處理,裡面的格式是:

testset.fasta是對蛋白質集合批處理,裡面的格式是(也可以單獨蛋白質存為.fasta文件,由於blast只能處理單條蛋白糊,把這個集合知識歸總的意思,第一步還是要生成單條蛋白質的.fasta文件,所以這個文件看個人意願):
import os
os.system('psiblast -query dataset/P00269.fasta -db /PSSM/uniref90.fasta -num_iterations 3 -out_ascii_pssm /dataset/P00269.pssm')##這個蛋白質好慢呀
import os
file_name='/dataset/testset.fasta'
Protein_id=[]
with open(file_name,'r') as fp:
i=0
for line in fp:
if i%2==0:
# Protein_id.append(line[1:-1])
id=line[0:-1]
p=line[1:-1]
with open ('/dataset/'+str(p)+'.fasta','a') as protein:
protein.write(id)
# protein.write()
if i%2==1:
seq=line[0:-1]
with open ('/dataset/'+str(p)+'.fasta','a') as protein:
protein.write('\n')
protein.write(seq)
i=i+1
os.system('psiblast -query '+'/dataset/'+str(p)+'.fasta -db /PSSM/uniref90.fasta -num_iterations 3 -out_ascii_pssm /dataset/'+str(p)+'.pssm')
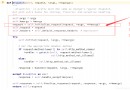
##PSSM真是太慢了,下面是只生成一個後的截圖
emmmm,在研究怎麼把這個矩陣存入文件方便調用,今天應該會更新……但是他好慢啊,不想用了。
參考文獻:
linux下用psiblast批量生成pssm矩陣_褚駿逸的博客-CSDN博客