introduction
PyVCF The installation of the library
PyVCF Library import
PyVCF Library details
Using examples :
_Record object ------ Storage form of location information
Reader object ------ Handle vcf file , Building structured information
Comprehensive use :
introductionvcf The full name of the document is variant call file, The mutation identification file , It's a document produced in the genome workflow , What's preserved is information about mutations in the genome . Through to vcf Document analysis , We can get individual variation information . Um. , All in all , This is a very important document , So how to deal with it is also very important . Its file information is as follows :
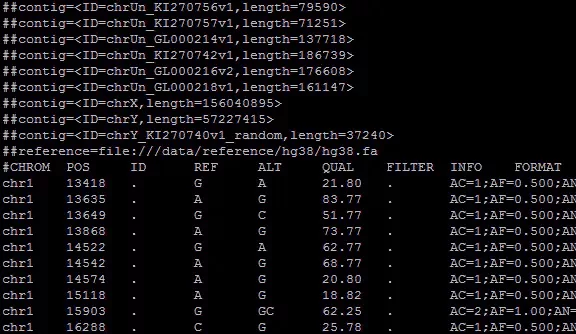
The beginning of the file is a pile of “##” Comment line at the beginning , Contains the basic information of the file . And then with “#” The first line , common 9+n Parts of , The first nine parts are marked with the information represented by each part in the following line , Equivalent to header . The following part is the sample name , There can be multiple . After the end of the comment line is the specific mutation information , Each line is divided into 9+n Parts of , Use tabs between each part (‘\t’) Separate .
Usually deal with vcf When you file , In the reading , There is always a lot of repetitive code written in the processing stage , The core task code is very few . Of course , If it's just looking for sites CHROM,POS,ID,REF,ALT,QUAL These parameters , You can do that . because vcf Format specification , The structure of these parameters is relatively simple . But if you deal with header information , Or deal with INFO,FORMAT When parameters are , To write more complex regular expressions , This is not only cumbersome , And it's easy to make mistakes .
Python Of PyVCF Library solves this problem , It uses regular expressions to put vcf File information is transformed into structured information , To simplify the vcf File processing , Facilitate the subsequent extraction of relevant parameters and processing .
PyVCF The installation of the librarycmd Interface
pip install PyVCFOr from https://github.com/jamescasbon/PyVCF Download the installation package from the website , Its installation .
PyVCF Library importimport vcfPyVCF The name of the library is vcf, After importing, you can use its method to vcf File processing .
PyVCF Library details Using examples :>>> import vcf>>> vcf_reader = vcf.Reader(filename=r'D:\test\example.hc.vcf.gz')>>> for record in vcf_reader: print recordRecord(CHROM=chr1, POS=10146, REF=AC, ALT=[A])Record(CHROM=chr1, POS=10347, REF=AACCCT, ALT=[A])Record(CHROM=chr1, POS=10439, REF=AC, ALT=[A])Record(CHROM=chr1, POS=10492, REF=C, ALT=[T])Record(CHROM=chr1, POS=10583, REF=G, ALT=[A])call vcf.Reader Class processing vcf file ,vcf The file information is saved to vcf_reader It's in . It's an iterative object , Its iteration element is a _Record Instance of object , Save a line of information that is not a comment line , That is, the specific information of the heterotopic point . Through it , We can easily get the details of the site .
_Record object ------ Storage form of location informationclass vcf.model._Record(CHROM, POS, ID, REF, ALT, QUAL, FILTER, INFO, FORMAT, sample_indexes, samples=None)_Record yes vcf.model One of the objects in , Besides it, there are _Call,_AltRecord Objects such as . Its basic attribute is CHROM,POS,ID,REF,ALT,QUAL,FILTER,INFO,FORMAT, That is to say vcf A line of site information in . Next, we will explain these attributes one by one :
CHROM: Chromosome name , The type is str.
POS: The position of the locus on the chromosome , The type is int.
ID: Usually mutated rs Number , The type is str. If it is ‘.’, Then for None.
REF: The base of the reference genome at this site , The type is str.
ALT: Sequencing results at this site . yes _AltRecord A list of subclass instances of the class . The type is list._AltRecord Like 4 A subclass , Represents several types of mutations : Such as snp,indel,structual variants etc. . All examples can be compared ( Limited to equal comparisons , There is no greater than less than ), Some subclasses do not implement str Method , In other words, it cannot be converted into a string .
QUAL: The sequencing quality of this site , The type is int or float.
FILTER: Filtering information . take FILTER A list of strings separated by semicolons , The type is list. If no parameters are given, it is None.
INFO: Some test indicators of this locus . take ‘=’ The previous parameter is used as the key , The following parameters are used as values , Build into a dictionary . The type is dict.
FORMAT: Genotype information . preservation vcf Of FORMAT Original form of column , The type is str.
>>> for record in vcf_reader: print type(record.CHROM), record.CHROM print type(record.POS), record.POS print type(record.ID), record.ID print type(record.REF), record.REF print type(record.ALT), record.ALT print type(record.QUAL), record.QUAL print type(record.FILTER), record.FILTER print type(record.INFO), record.INFO print type(record.INFO['BaseQRankSum']), record.INFO['BaseQRankSum'] print type(record.FORMAT), record.FORMAT <type 'str'> chr1<type 'int'> 234481<type 'NoneType'> None<type 'str'> T<type 'list'> [A]<type 'float'> 2025.77<type 'NoneType'> None<type 'dict'> {'ExcessHet': 3.0103, 'AC': [1], 'BaseQRankSum': -2.743, 'MLEAF': [0.5], 'AF': [0.5], 'MLEAC': [1], 'AN': 2, 'FS': 2.371, 'MQ': 42.83, 'ClippingRankSum': 0.0, 'SOR': 0.972, 'MQRankSum': -2.408, 'ReadPosRankSum': 1.39, 'DP': 156, 'QD': 13.07}<type 'float'>-2.743<type 'str'>GT:AD:DP:GQ:PLIn addition to these basic attributes ,_Record Objects have some other properties :
samples: hold FORMAT Information as a key , The following corresponding information is taken as the value , Build into a dictionary (CallData object ), as well as sample name , These two values form a Call object , Co constitute samples An element of . This way sample And genotype information , Press the subscript to access each Call object .samples The type is list.
start: Where the mutation started
end: Where the mutation ends
alleles: All possible conditions of this site , from REF and ALT A list of parameters (REF The type is str,ALT Parameter is _AltRecord Subclass instances of objects ), The type is list.
>>> for record in vcf_reader: print record.samples, '\n', record.samples[0].sample, '\n', record.samples[0]['GT'] # Access by subscript Call, Press .sample visit sample, Press to access FORMAT Corresponding information print record.start, record.POS, record.end print record.REF, record.ALT, record.alleles # Be careful G No quotes , It is _AltRecord object [Call(sample=192.168.1.1, CallData(GT=0/1, AD=[39, 14], DP=53, GQ=99, PGT=0|1, PID=13116_T_G, PL=[449, 0, 2224]))] 192.168.1.10/113115 13116 13116T [G] ['T', G]_Record Object methods :
Compare size methods between objects : Compare according to chromosome name and location information .Python3 only ‘=’ and ‘<’ Comparison .
Iterative method : Yes samples Iterate over the elements in .
String method : Only return CHROM,POS,REF,ALT Four columns of information .
genotype(name) Method , and samples Press subscript to access different , This method provides by sample Name the function to access .
add_format(fmt), add_filter(flt), add_info(info, value=True): Add elements to the corresponding attributes .
get_hom_refs(): Get samples All that are not mutated at this site sample, Returns a list of .
get_hom_alts(): Get samples At this site 100% All of the mutations sample, Returns a list of .
get_hets(): Get samples All genotypes at this locus are heterozygous sample, Returns a list of .
get_unknown(): Get samples In all cases where the genotype of this locus is unknown sample, Returns a list of .
>>> record = next(vcf_reader)>>> record2 = next(vcf_reader)>>> print record > record2 # Compare by chromosome name and location False>>> for i in record: # Press samples The list iterates print i Call(sample=192.168.1.1,CallData(GT=0/1, AD=[18, 11], DP=29, GQ=99, PL=[280, 0, 528]))>>> print str(record) # String method Record(CHROM=chr1, POS=10492, REF=C, ALT=[T])>>> print record.genotype('192.168.1.1') # Press sample Name to access Call(sample=192.168.1.1, CallData(GT=0/1, AD=[39, 14], DP=53, GQ=99, PGT=0|1, PID=13116_T_G, PL=[449, 0, 2224]))_Record Objects also have many useful method properties :
num_called: The site has been identified sample number .
call_rate: Identified sample Number accounting for sample The proportion of the total .
num_hom_ref,num_hom_alt,num_het,num_unknown: The number of four genotypes
aaf: all sample Allele frequency ( Except REF), Returns a list of .
heterozygosity: Heterozygosity of this site ,0.5 Heterozygous mutation ,0 Homozygous mutation .
var_type: Mutation types , Include ‘snp’,‘indel’,‘sv’(structural variant),‘unknown’.
var_subtype: More refined mutation types , Such as ‘indel’ Include ‘del’,‘ins’,‘unknown’.
is_snp,is_indel,is_sv,is_transition,is_deletion: Judge whether the mutation is snp,indel,sv,transition,deletion wait .
>>> record = next(vcf_reader)>>> print recordRecord(CHROM=chr1, POS=13118, REF=A, ALT=[G])>>> print record.samples # only one sample[Call(sample=192.168.1.1, CallData(GT=0/1, AD=[41, 13], DP=54, GQ=99, PGT=0|1, PID=13116_T_G, PL=[449, 0, 2224]))]>>> record.num_called1>>> record.call_rate1.0>>> record.num_hom_ref0>>> record.aaf[0.5]>>> record.num_het1>>> record.heterozygosity0.5>>> record.var_type'snp'>>> record.var_subtype'ts'>>> record.is_snpTrue>>> record.is_indelFalseReader object ------ Handle vcf file , Building structured information class Reader(fsock=None, filename=None, compressed=None, prepend_chr=False, strict_whitespace=False, encoding='ascii')reading vcf When you file , There are a total of six parameters to choose from , As shown in the figure above .
fsock: The file object of the target file , It can be used open( file name ) Get this file object .
filename: file name , When fsock and filename Simultaneous existence , Give priority to fsock.
compressed: Do you want to decompress , If no parameters are provided, the program will judge by itself ( Whether the file name is .gz At the end, judge whether you need to decompress ).
prepend_chr: When saving chromosome names , Prefix or not ‘chr’, The default without , If vcf The chromosome name of the file does not have a prefix ‘chr’, Can be set to True, Automatically add .
strict_whitespace: Is it strictly tab ‘\t’ Separate data .True Then it means strictly according to tab ,False Indicates that spaces can be mixed .
encoding: File encoding .
>>> vcf_reader = vcf.Reader(open('vcf/test/example-4.0.vcf', 'r')) #fsock>>> vcf_reader = vcf.Reader(filename=r'D:\test\example.hc.vcf.gz') #filenameHeader information is mainly stored in Reader In the properties of the object , Include alts,contigs,filters,formats,infos,metadata.
alts Using examples :
>>> vcf_reader = vcf.Reader(filename=r'D:\test\example.hc.vcf.gz')>>> vcf_reader.altsOrderedDict([('NON_REF', Alt(id='NON_REF', desc='Represents any possible alternative allele at this location'))]) # Dictionary type >>> vcf_reader.alts['NON_REF'].id'NON_REF'>>> vcf_reader.alts['NON_REF'].desc'Represents any possible alternative allele at this location'The usage of other attributes is similar .
Reader Object implements two methods :
next(): Get the data of the next row , That is to return to the next _Record object . Can be explicitly called next() Get the next row of data , You can also iterate directly Reader object , It will automatically call next() Function to get the next row of data .
fetch(chrom,start=None,end=None): return chrom Chromosomes from start+1 To end All mutation sites of coordinates . Do not give end, Just go back to chrom Chromosomes from start+1 All mutation sites to the end ;
start and end Not even , Just go back to chrom All mutation sites on chromosome . This method requires another third party Python modular pysam To index files , If this module is not installed , Can cause errors .
in addition , After using this method , It will change the iteratable range of the object to fetch() The obtained mutation site , So use this method , The original iteration schedule is invalid .
>>> vcf_reader = vcf.Reader(filename=r'D:\test\example.hc.vcf.gz')>>> vcf_reader.next()<vcf.model._Record object at 0x0000000003ED8780>>>> record = vcf_reader.next()>>> print recordRecord(CHROM=chr1, POS=10347, REF=AACCCT, ALT=[A])>>> for record in vcf_reader: print recordRecord(CHROM=chr1, POS=10439, REF=AC, ALT=[A])Record(CHROM=chr1, POS=10492, REF=C, ALT=[T])This library has another Writer object , I won't go into details here , Because most of them are right vcf The processing of documents can be done with the knowledge of the above two objects .
Comprehensive use :#!/usr/bin/env python# -*- coding: utf-8 -*- import vcf # Import PyVCF library filename = r'D:\test\example.hc.vcf.gz' vcf_reader = vcf.Reader(filename=filename) # call Reader Object processing vcf file for record in vcf_reader: # iteration Reader object , The return is _Record object # record yes _Record object print record.CHROM, record.POS, record.ID, record.ALT if record.is_snp:# Judge whether it is snp print "I'm a snp" elif record.var_type != 'sv': # and elif record.is_sv: Equivalent print "I'm not a sv" if record.heterozygosity == 0.5: # Determine whether it is a heterozygous mutation print "I'm a heterozygous mutation" ... ...All the functions implemented by this library , Can write their own code to achieve , And the implementation method is relatively simple . The reason why we use this library to deal with vcf file , Because this library may consider more things than we know ourselves , Its implementation may also be more complete and reasonable than our own code .
also , It's always bad to build cars repeatedly .
That's all python PyVCF Document processing VCF Details of file format examples , More about python PyVCF Handle VCF Please pay attention to other relevant articles of software development network for information in format !